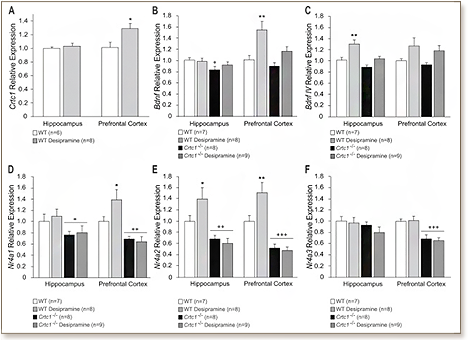
 Major depression is a highly complex disabling psychiatric disorder affecting millions of people worldwide. Despite the availability of several classes of antidepressants, a substantial percentage of patients are unresponsive to these medications. A better understanding of the neurobiology of depression and the mechanisms underlying antidepressant response is thus critically needed. We previously reported that mice lacking CREB-regulated transcription coactivator 1 (CRTC1) exhibit a depressive-like phenotype and a blunted antidepressant response to the selective serotonin reuptake inhibitor fluoxetine. In this study, we similarly show that Crtc1-/- mice are resistant to the antidepressant effect of chronic desipramine in a behavioral despair paradigm. Supporting the blunted response to this tricyclic antidepressant, we found that desipramine does not significantly increase the expression of Bdnf and Nr4a1-3 in the hippocampus and prefrontal cortex of Crtc1-/- mice. Epigenetic regulation of neuroplasticity gene expression has been associated with depression and antidepressant response, and histone deacetylase (HDAC) inhibitors have been shown to have antidepressant-like properties. Here, we show that unlike conventional antidepressants, chronic systemic administration of the HDAC inhibitor SAHA partially rescues the depressive-like behavior of Crtc1-/- mice. This behavioral effect is accompanied by an increased expression of Bdnf, but not Nr4a1-3, in the prefrontal cortex of these mice, suggesting that this epigenetic intervention restores the expression of a subset of genes by acting downstream of CRTC1. These findings suggest that CRTC1 alterations may be associated with treatment-resistant depression, and support the interesting possibility that targeting HDACs may be a useful therapeutic strategy in antidepressant development.
Major depression is a highly complex disabling psychiatric disorder affecting millions of people worldwide. Despite the availability of several classes of antidepressants, a substantial percentage of patients are unresponsive to these medications. A better understanding of the neurobiology of depression and the mechanisms underlying antidepressant response is thus critically needed. We previously reported that mice lacking CREB-regulated transcription coactivator 1 (CRTC1) exhibit a depressive-like phenotype and a blunted antidepressant response to the selective serotonin reuptake inhibitor fluoxetine. In this study, we similarly show that Crtc1-/- mice are resistant to the antidepressant effect of chronic desipramine in a behavioral despair paradigm. Supporting the blunted response to this tricyclic antidepressant, we found that desipramine does not significantly increase the expression of Bdnf and Nr4a1-3 in the hippocampus and prefrontal cortex of Crtc1-/- mice. Epigenetic regulation of neuroplasticity gene expression has been associated with depression and antidepressant response, and histone deacetylase (HDAC) inhibitors have been shown to have antidepressant-like properties. Here, we show that unlike conventional antidepressants, chronic systemic administration of the HDAC inhibitor SAHA partially rescues the depressive-like behavior of Crtc1-/- mice. This behavioral effect is accompanied by an increased expression of Bdnf, but not Nr4a1-3, in the prefrontal cortex of these mice, suggesting that this epigenetic intervention restores the expression of a subset of genes by acting downstream of CRTC1. These findings suggest that CRTC1 alterations may be associated with treatment-resistant depression, and support the interesting possibility that targeting HDACs may be a useful therapeutic strategy in antidepressant development.